Chemical potential for SPC/E water at 1 atm and 300 K
- hmcezar
-
 Topic Author
Topic Author
- Offline
- New Member
-

Less
More
- Posts: 4
- Thank you received: 0
3 years 9 months ago #744
by hmcezar
Hello everyone,
I'm performing GCMC simulations of liquid water using the SPC/E force field.
What I want is to have a simulation running at around 1 atm and about 998 g/l at 300 K.
I saw in the literature that for the SPC/E force field, these conditions for the simulation are given when the chemical potential is -38.09 ( dx.doi.org/10.1063/1.5046835 ).
I tried using this value in Cassandra without success, since the simulations converge to huge pressures (average 6248 stddev 551, from a equilibration run with about 10^8 steps, which is not much, but I believe it gives the trend).
Doing some tests, I got that a chemical potential of about -48.25 gives me something close to the thermodynamic conditions I want.
From Cassandra's paper I know that a shifted chemical potential is used, but I thought it was just internally.
Since it seems that it's not the case, and that the shifted chemical potential is the parameters given by the user, I'd like to know:
1) Is there any formula to convert between the shifted chemical potential and the chemical potential that I got from the literature? I did not find this expression in the manual.
2) In case there's not, how do I know that the chemical potential of -48.25 gives me the condition that I want. I mean, how do I know it's not -48.2, or -48.3, for instance. I ask because since it's a simulation of a liquid, the pressures changes a lot and the standard deviations are in the orders of magnitude of the pressure itself (average 231, stddev 160). Do I really need to run simulations with more than 10^10 steps just to get an idea of the values?
Regards,
Henrique
I'm performing GCMC simulations of liquid water using the SPC/E force field.
What I want is to have a simulation running at around 1 atm and about 998 g/l at 300 K.
I saw in the literature that for the SPC/E force field, these conditions for the simulation are given when the chemical potential is -38.09 ( dx.doi.org/10.1063/1.5046835 ).
I tried using this value in Cassandra without success, since the simulations converge to huge pressures (average 6248 stddev 551, from a equilibration run with about 10^8 steps, which is not much, but I believe it gives the trend).
Doing some tests, I got that a chemical potential of about -48.25 gives me something close to the thermodynamic conditions I want.
From Cassandra's paper I know that a shifted chemical potential is used, but I thought it was just internally.
Since it seems that it's not the case, and that the shifted chemical potential is the parameters given by the user, I'd like to know:
1) Is there any formula to convert between the shifted chemical potential and the chemical potential that I got from the literature? I did not find this expression in the manual.
2) In case there's not, how do I know that the chemical potential of -48.25 gives me the condition that I want. I mean, how do I know it's not -48.2, or -48.3, for instance. I ask because since it's a simulation of a liquid, the pressures changes a lot and the standard deviations are in the orders of magnitude of the pressure itself (average 231, stddev 160). Do I really need to run simulations with more than 10^10 steps just to get an idea of the values?
Regards,
Henrique
Please Log in to join the conversation.
- ryangmullen
-
- Offline
- Administrator
-

Less
More
- Posts: 124
- Karma: 4
- Thank you received: 24
3 years 9 months ago #745
by ryangmullen
Hi Henrique,
The relationship between the chemical potential and the shifted chemical potential is given as Eq 7.31 in the Cassandra user guide. In general, the four partition functions you need to convert between the two are only knowable for a few special cases. For monatomic molecules, each partition function reduces to unity, and the chemical potential isn't shifted. For a rigid, non-linear molecule like SPC/E water:
Ryan
The relationship between the chemical potential and the shifted chemical potential is given as Eq 7.31 in the Cassandra user guide. In general, the four partition functions you need to convert between the two are only knowable for a few special cases. For monatomic molecules, each partition function reduces to unity, and the chemical potential isn't shifted. For a rigid, non-linear molecule like SPC/E water:
- Omega.dih = 1
- Z.frag = Z.int = 1
- Q.rot+int = Q.rot Q.int = Q.rot
Ryan
Please Log in to join the conversation.
- hmcezar
-
Topic Author
- Offline
- New Member
-
Less
More
- Posts: 4
- Thank you received: 0
3 years 9 months ago - 3 years 9 months ago #746
by hmcezar
Hi Ryan,
Thank you very much for pointing me out to the right direction.
Somehow I've missed the equation in the user guide, sorry about that.
Anyways, I followed your suggestion and got the Qrot term from McQuarrie.
I did the math more than once, but after converting the chemical potential from the reference I gave in the first post, I get a shifted chemical potential of -28.682 kJ/mol.
Since this value is greater than the chemical potentials I tested and got huge pressures, it is expected that I'll get even higher pressures.
This is the expression I used for Qrot:
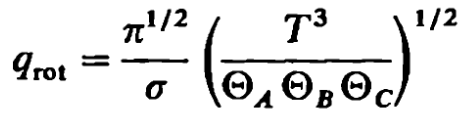
with \Theta_A = 40.1 K, \Theta_B = 20.9 K, \Theta_C = 13.4 K, as provided in Table 8-1 of McQuarrie for H2O.
Using a temperature of 300 K I get Qrot = 43.4539334 which leads me to value of 9.4079 that should be added to the chemical potential to get the shifted chemical potential, hence the value of -28.682 kJ/mol.
Did I miss something?
Regards,
Henrique
Thank you very much for pointing me out to the right direction.
Somehow I've missed the equation in the user guide, sorry about that.
Anyways, I followed your suggestion and got the Qrot term from McQuarrie.
I did the math more than once, but after converting the chemical potential from the reference I gave in the first post, I get a shifted chemical potential of -28.682 kJ/mol.
Since this value is greater than the chemical potentials I tested and got huge pressures, it is expected that I'll get even higher pressures.
This is the expression I used for Qrot:
with \Theta_A = 40.1 K, \Theta_B = 20.9 K, \Theta_C = 13.4 K, as provided in Table 8-1 of McQuarrie for H2O.
Using a temperature of 300 K I get Qrot = 43.4539334 which leads me to value of 9.4079 that should be added to the chemical potential to get the shifted chemical potential, hence the value of -28.682 kJ/mol.
Did I miss something?
Regards,
Henrique
Last Edit: 3 years 9 months ago by hmcezar. Reason: Even though I double checked, I did the math wrong, now I believe it's correct.
Please Log in to join the conversation.
- ryangmullen
-
- Offline
- Administrator
-
Less
More
- Posts: 124
- Karma: 4
- Thank you received: 24
3 years 9 months ago - 3 years 9 months ago #747
by ryangmullen
Reading over the paper from which you pulled the chemical potential, Belloni gives this detail on pg 2:
"The value of µ is found to be around −15.37kT = −38.10 kJ/mol where the activity a = exp(β µ) is expressed in Å−3 (or, equivalently, when the de Broglie length is set at 1 Å and the rotational kinetic function is ignored in the complete expression of µ)"
In other words, the µ Belloni reports has been shifted by both the rotational partition function term, +RT ln(Q.rot), and by the de Broglie wavelength term, -3RT ln(Λ):
µ.Belloni = µ + RT ln(Q.rot) - 3RT ln(Λ)
The chemical potential that Cassandra requires needs only be shifted by +RT ln(Q.rot), since Cassandra computes Λ from the mass of the molecule (0.2384 Å for water at 300K) and includes it in the acceptance criteria:
µ.Cassandra = µ + RT ln(Q.rot)
If you want to run simulations in Cassandra at the same conditions Belloni reports, then
µ.Cassandra = µ.Belloni + 3RT ln(Λ) = -38.1 -10.7 kJ/mol = -48.8 kJ/mol
"The value of µ is found to be around −15.37kT = −38.10 kJ/mol where the activity a = exp(β µ) is expressed in Å−3 (or, equivalently, when the de Broglie length is set at 1 Å and the rotational kinetic function is ignored in the complete expression of µ)"
In other words, the µ Belloni reports has been shifted by both the rotational partition function term, +RT ln(Q.rot), and by the de Broglie wavelength term, -3RT ln(Λ):
µ.Belloni = µ + RT ln(Q.rot) - 3RT ln(Λ)
The chemical potential that Cassandra requires needs only be shifted by +RT ln(Q.rot), since Cassandra computes Λ from the mass of the molecule (0.2384 Å for water at 300K) and includes it in the acceptance criteria:
µ.Cassandra = µ + RT ln(Q.rot)
If you want to run simulations in Cassandra at the same conditions Belloni reports, then
µ.Cassandra = µ.Belloni + 3RT ln(Λ) = -38.1 -10.7 kJ/mol = -48.8 kJ/mol
Last Edit: 3 years 9 months ago by ryangmullen.
Please Log in to join the conversation.
Time to create page: 0.169 seconds